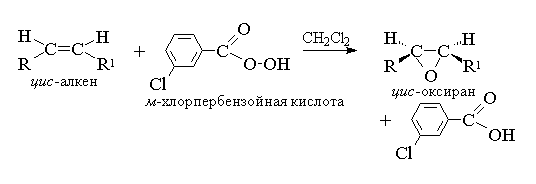
Окисление алкенов (ациклических и циклических) при взаимодействии с перкислотами (надкислотами) в неполярной, индифферентной среде сопровождается образованием окисей алкенов – эпоксиды, поэтому сама реакция носит название реакции эпоксидирования.
Согласно современно номенклатуре ИЮПАК, трехчленный цикл с одним атомом кислорода носит название оксиран.
Эпоксидирование алкенов следует рассматривать как синхронный, согласованный процесс, в котором не участвуют ионные интермедиаты типа гидроксильного катиона OH+. Эпоксидирование алкенов представляет собой процесс син-присоединения одного атома кислорода по двойной связи с полным сохранением конфигурации заместителей при двойной связи:
Для эпоксидирования был предложен механизм, характерный для согласованных процессов:
В качестве эпоксидирующих агентов используются перкислоты: пербензойная, м-хлорпербензойная, мононадфталевая, перуксусная, пертрифторуксусная и пермуравьиная. Перкислоты ароматического ряда применяют в виде индивидуальных реагентов, тогда как перкислоты алифатического ряда – CH3CO3H, CF3CO3H и HCO3H – не выделяют индивидуально и используют сразу после их образования при взаимодействии 30- или 90%-й перекиси водорода и соответствующей карбоновой кислоты. Пербензойную и мета-хлорпербензойную кислоты в настоящее время получают окислением соответственно бензойной и мета-хлорбензойной кислот 70%-й перкисью водорода в растворе метансульфокислоты:
или из хлорангидридов кислот и перекиси водорода:
Мононадфталевую кислоту получают подобным методом из фталевого ангидрида и 30%-й перекиси водорода в водной щелочи:
Первоначально для получения оксиранов (эпоксидов) использовалась пербензойная или мононадфталевая кислоты:
Особенно удобен метод с использованием мононадфталевой кислоты. Мононадфталевая кислота хорошо растворима в эфире, тогда как один из продуктов реакции (фталевая кислота) совершенно не растворим в эфире, и о ходе реакции легко судить по количеству выделившейся кристаллической фталевой кислоты.
В настоящее время для эпоксидирования чаще всего используют мета-хлорпербензойную кислоту. В отличие от других перкислот она стабильная при хранении в течение длительного времени (до 1 года) и абсолютно безопасная при обращении. Выходы оксиранов, полученных при окислении ациклических и циклических алкенов мета-хлорпербензойной кислотой в растворе хлористого метилена, обычно очень высокие.
Перкислоты часто генерируют прямо в реакционной смеси из 90%-й перекиси водорода и карбоновой кислоты в хлористом метилене:
Алкены, с двойной связью, сопряженной с карбонильной и карбоксильной группой или другим акцепторным заместителем, малоактивны, и для их окисления необходимо использовать более сильные окислители, такие как трифторперуксусную кислоту, получаемую из ангидрида трифторуксусной кислоты и 90%-й перекиси водорода в хлористом метилене. Альтернативный метод эпоксидирования заключается во взаимодействии алкена с нитрилом и 90%-й перекисью водорода:
Простейший оксиран – окись этилена – получают в промышленности окислением этилена кислородом в присутствии серебра как катализатора:
Трехчленное кольцо оксиранов легко раскрывается под действием самых разнообразных нуклеофильных реагентов. Эти реакции подробно будут обсуждаться в главе 11, посвященной ациклическим и циклическим простым эфирам. Здесь же будет рассмотрен только гидролиз эпоксидов. Гидролиз эпоксидов катализируется как кислотами, так и основаниями. В обоих случаях образуются вицинальные диолы, т.е. гликоли. При кислотном катализе в первой стадии происходит протонирование атома кислорода эпоксида с образованием циклического оксониевого иона, который раскрывается в результате нуклеофильной атаки молекулы воды:
Ключевой стадией в раскрытии кольца, определяющей скорость всего процесса, является нуклеофильная атака водой на протонированную форму эпоксида. С точки зрения механизма этот процесс аналогичен раскрытию бромониевого иона при нуклеофильной атаке бромид-иона или другого нуклеофильного агента. С этих позиций стереохимическим результатом должно быть образование транс-гликолей при расщеплении циклических эпоксидов. Действительно, при кислотно-катализируемом гидролизе циклогексеноксида или циклопентеноксида образуются исключительно транс-1,2-диолы:
Таким образом, двухстадийный процесс эпоксидирования алкена с последующим кислотным гидролизом эпоксида суммарно соответствует реакции анти-гидроксилирования алкенов.
Обе стадии анти-гидроксилирования алкенов можно совместить, если алкен обрабатывать водной 30 – 70%-й перекисью водорода в муравьиной или трифторуксусной кислоте. Обе эти кислоты являются достаточно сильными для того, чтобы вызвать раскрытие эпоксидного цикла, поэтому их обычно используют для анти-гидроксилирования алкенов, например:
Раскрытие эпоксидного кольца, катализируемое основанием, также приводит к образованию транс-гликолей:
Следовательно, двухстадийный процесс эпоксидирования алкенов с последующим щелочным гидролизом эпоксидов также является реакцией анти-гидроксилирования алкенов.
Третий современный метод анти-гидроксилирования алкенов был предложен и разработан К. Прево (1933 г.). Алкен нагревают с йодом и бензоатом или ацетатом серебра в безводном бензоле или CCl4. транс-Присоединение к двойной связи первоначально приводит к образованию йодэфира, в котором йод далее замещается бензоат-ионом, и получается дибензоат гликоля:
Реакция Прево в безводной среде приводит к образованию того же диола, что и эпоксидирование алкенов с последующим гидролизом:
Таким образом, реакция Прево представляет собой более дорогостоящую модификацию других методов анти-гидроксилирования алкенов. Однако для чувствительных к действию кислот соединений этот метод имеет очевидные преимущества перед методом анти-гидроксилирования с помощью перкислот и последующего кислотного гидролиза эпоксида.
Некоторые соли и оксиды переходных металлов высших степенях окисления являются эффективными реагентами син-гидроксилирования двойной связи. Окисление алкенов перманганатом калия – один из старейших методов син-гидроксилирования двойной связи – продолжает широко использоваться несмотря на свойственные ему ограничения. цис-1,2-Циклогександиол был впервые получен В.В. Марковниковым еще в 1878 г. гидроксилированием циклогексена водным раствором перманганата калия при 0ºС:
Этот метод в дальнейшем получил развитие в работах русского ученого Е.Е, Вагнера, поэтому син-гидроксилирование под действием водного раствора перманганата калия носит название реакции Вагнера. Перманганат калия является сильным окислителем, способным е только гидроксилировать двойную связь, но и расщеплять образующийся вицинальный диол. Чтобы по возможности избежать дальнейшего расщепления гликолей, необходимо тщательно контролировать условия реакции. Наилучшие результаты достигаются при гидроксилировании алкенов в слабощелочной среде (pH ~ 8) при 0 – 5ºС разбавленным ~ 1% водным раствором KMnO4. Тем не менее выходы гликолей обычно невелики (30 – 60%):
Первоначально при окислении алкенов перманганатом калия образуется циклический эфир марганцевой кислоты, который немедленно гидролизуется до вицинального диола:
Циклический эфир марганцевой кислоты как интермедиат никогда не был выделен, однако его образование следует из экспериментов с меченым 18O перманганатом калия. К. Вайберг с сотрудниками (1957 г.) показали, что оба атома кислорода в гликоле оказываются мечеными при окислении алкена KMn18O4. Это означает, что оба атома кислорода переходят от окислителя, а не из растворителя – воды, что находится в хорошем соответствии с предлагаемым механизмом.
Другой метод син-гидроксилирования алкенов под действием оксида осмия (VIII) OsO4 был предложен Р. Криге в 1936 г. Тетраоксид осмия представляет собой бесцветное кристаллическое вещество, хорошо растворимое в эфире, диоксане, пиридине и других органических растворителях. При взаимодействии тетраоксида осмия с алкенами в эфире или диоксане образуется черный осадок циклического эфира осмиевой кислоты – осмат, который легко может быть изолирован в индивидуальном виде. Присоединение OsO4 к двойной связи заметно ускоряется в растворе пиридина. Разложение осматов до вицинальных диолов достигается действием водного раствора гидросульфита натрия или сероводородом:
Выходы продуктов син-гидроксилирования алкенов в этом методе значительно выше, чем при использовании перманганата в качестве окислителя. Важным достоинством метода Криге является отсутствие продуктов окислительного расщепления алкенов, характерного для перманганатного окисления:
Тетраоксид осмия – дорогой и труднодоступный реагент, к тому же он очень токсичен. Поэтому оксид осмия (VIII) используют для синтеза малых количеств труднодоступных веществ с целью получения наиболее высокого выхода диола. Для упрощения син-гидроксилирования алкенов под действием OsO4 была разработана методика, позволяющая использовать лишь каталитические количества этого реагента. Гидроксилирование осуществляется с помощью перекиси водорода в присутствии OsO4, например:
Интересно отметить, что высшие оксиды других переходных металлов (V2O5, WO3, MoO3 и др.) катализируют анти-гидроксилирование алкенов.
Р. Вудворд в 1958 г. предложил альтернативный трехстадийный способ син-гидроксилирования алкенов. Первоначально алкен превращают в транс-йодацетат в результате взаимодействия с йодом и ацетатом серебра в уксусной кислоте. Затем галоген замещаю на оксигрупу при обработке водной уксусной кислотой при нагревании. Последняя стадия заключается в гидролитическом отщеплении ацетатной группы:
В заключение этого раздела приведем стереохимические отношения между алкеном цис- или транс-конфигурации и конфигурацией образующегося вицинального гликоля, который может быть цис- или транс-изомером, эритро- или трео-формой, мезо- или d-,l-формой, в зависимости от заместителей в алкене:
Аналогичные стереохимические отношения наблюдаются и в других реакциях син- или анти-присоединения водорода, галогеноводородов, воды, галогенов, гидридов бора и других реагентов по кратной связи.
В окислительно-восстановительных реакциях органические вещества чаще проявляют свойства восстановителей, а сами окисляются. Легкость окисления органических соединений зависит от доступности электронов при взаимодействии с окислителем. Все известные факторы, вызывающие увеличение электронной плотности в молекулах органических соединений (например, положительные индуктивный и мезомерные эффекты), будут повышать их способность к окислению и наоборот.
Склонность органических соединений к окислению возрастает с ростом их нуклеофильности , что соответствует следующим рядам:
Рост нуклеофильности в ряду
Рассмотрим окислительно-восстановительные реакции представителей важнейших классов органических веществ с некоторыми неорганическими окислителями.
Окисление алкенов
При мягком окислении алкены превращаются в гликоли (двухатомные спирты). Атомы-восстановители в этих реакциях – атомы углерода, связанные двойной связью.
Реакция с раствором перманганата калия протекает в нейтральной или слабо щелочной среде следующим образом:
3C 2 H 4 + 2KMnO 4 + 4H 2 O → 3CH 2 OH–CH 2 OH + 2MnO 2 + 2KOH
В более жестких условиях окисление приводит к разрыву углеродной цепи по двойной связи и образованию двух кислот (в сильно щелочной среде – двух солей) или кислоты и диоксида углерода (в сильно щелочной среде – соли и карбоната):
1) 5CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5C 2 H 5 COOH + 8MnSO 4 + 4K 2 SO 4 + 17H 2 O
2) 5CH 3 CH=CH 2 + 10KMnO 4 + 15H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 10MnSO 4 + 5K 2 SO 4 + 20H 2 O
3) CH 3 CH=CHCH 2 CH 3 + 8KMnO 4 + 10KOH → CH 3 COOK + C 2 H 5 COOK + 6H 2 O + 8K 2 MnO 4
4) CH 3 CH=CH 2 + 10KMnO 4 + 13KOH → CH 3 COOK + K 2 CO 3 + 8H 2 O + 10K 2 MnO 4
Дихромат калия в сернокислотной среде окисляет алкены аналогично реакциям 1 и 2.
При окислении алкенов, в которых атомы углерода при двойной связи содержат по два углеродных радикала, происходит образование двух кетонов:


Окисление алкинов
Алкины окисляются в несколько более жестких условиях, чем алкены, поэтому они обычно окисляются с разрывом углеродной цепи по тройной связи. Как и в случае алкенов, атомы-восстановители здесь – атомы углерода, связанные кратной связью. В результате реакций образуются кислоты и диоксид углерода. Окисление может быть проведено перманганатом или дихроматом калия в кислотной среде, например:
5CH 3 C≡CH + 8KMnO 4 + 12H 2 SO 4 → 5CH 3 COOH + 5CO 2 + 8MnSO 4 + 4K 2 SO 4 + 12H 2 O

Ацетилен может быть окислен перманганатом калия в нейтральной среде до оксалата калия:
3CH≡CH +8KMnO 4 → 3KOOC –COOK +8MnO 2 +2КОН +2Н 2 О
В кислотной среде окисление идет до щавелевой кислоты или углекислого газа:
5CH≡CH +8KMnO 4 +12H 2 SO 4 → 5HOOC –COOH +8MnSO 4 +4К 2 SO 4 +12Н 2 О
CH≡CH + 2KMnO 4 +3H 2 SO 4 → 2CO 2 + 2MnSO 4 + 4H 2 O + K 2 SO 4
Окисление гомологов бензола
Бензол не окисляется даже в довольно жестких условиях. Гомологи бензола могут быть окислены раствором перманганата калия в нейтральной среде до бензоата калия:
C 6 H 5 CH 3 +2KMnO 4 → C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O
C 6 H 5 CH 2 CH 3 + 4KMnO 4 → C 6 H 5 COOK + K 2 CO 3 + 2H 2 O + 4MnO 2 + KOH
Окисление гомологов бензола дихроматом или перманганатом калия в кислотной среде приводит к образованию бензойной кислоты.
5С 6 Н 5 СН 3 +6КMnO 4 +9 H 2 SO 4 → 5С 6 Н 5 СООН+6MnSO 4 +3K 2 SO 4 + 14H 2 O
5C 6 H 5 –C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 → 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O



Окисление спиртов
Непосредственным продуктом окисления первичных спиртов являются альдегиды, а вторичных – кетоны.
Образующиеся при окислении спиртов альдегиды легко окисляются до кислот, поэтому альдегиды из первичных спиртов получают окислением дихроматом калия в кислотной среде при температуре кипения альдегида. Испаряясь, альдегиды не успевают окислиться.
3C 2 H 5 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O
С избытком окислителя (KMnO 4 , K 2 Cr 2 O 7) в любой среде первичные спирты окисляются до карбоновых кислот или их солей, а вторичные – до кетонов.
5C 2 H 5 OH + 4KMnO 4 + 6H 2 SO 4 → 5CH 3 COOH + 4MnSO 4 + 2K 2 SO 4 + 11H 2 O
3CH 3 –CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CH 3 –COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
Третичные спирты в этих условиях не окисляются, а метиловый спирт окисляется до углекислого газа.

Двухатомный спирт, этиленгликоль HOCH 2 –CH 2 OH, при нагревании в кислой среде с раствором KMnO 4 или K 2 Cr 2 O 7 легко окисляется до щавелевой кислоты, а в нейтральной – до оксалата калия.
5СН 2 (ОН) – СН 2 (ОН) + 8КMnO 4 +12H 2 SO 4 → 5HOOC –COOH +8MnSO 4 +4К 2 SO 4 +22Н 2 О
3СН 2 (ОН) – СН 2 (ОН) + 8КMnO 4 → 3KOOC –COOK +8MnO 2 +2КОН +8Н 2 О
Окисление альдегидов и кетонов
Альдегиды – довольно сильные восстановители, и поэтому легко окисляются различными окислителями, например: KMnO 4 , K 2 Cr 2 O 7 , OH, Cu(OH) 2 . Все реакции идут при нагревании:
3CH 3 CHO + 2KMnO 4 → CH 3 COOH + 2CH 3 COOK + 2MnO 2 + H 2 O
3CH 3 CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 → 3CH 3 COOH + Cr 2 (SO 4) 3 + 7H 2 O
CH 3 CHO + 2KMnO 4 + 3KOH → CH 3 COOK + 2K 2 MnO 4 + 2H 2 O
5CH 3 CHO + 2KMnO 4 + 3H 2 SO 4 → 5CH 3 COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O
CH 3 CHO + Br 2 + 3NaOH → CH 3 COONa + 2NaBr + 2H 2 O

реакция «серебряного зеркала»
C аммиачным раствором оксида серебра альдегиды окисляются до карбоновых кислот которые в аммиачном растворе дают соли аммония (реакция «серебрянного зеркала»):
CH 3 CH=O + 2OH → CH 3 COONH 4 + 2Ag + H 2 O + 3NH 3
CH 3 –CH=O + 2Cu(OH) 2 → CH 3 COOH + Cu 2 O + 2H 2 O
Муравьиный альдегид (формальдегид) окисляется, как правило, до углекислого газа:
5HCOH + 4KMnO 4 (изб ) + 6H 2 SO 4 → 4MnSO 4 + 2K 2 SO 4 + 5CO 2 + 11H 2 O
3СН 2 О + 2K 2 Cr 2 O 7 + 8H 2 SO 4 → 3CO 2 +2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O
HCHO + 4OH → (NH 4) 2 CO 3 + 4Ag↓ + 2H 2 O + 6NH 3
HCOH + 4Cu(OH) 2 → CO 2 + 2Cu 2 O↓+ 5H 2 O
Кетоны окисляются в жестких условия сильными окислителями с разрывом связей С-С и дают смеси кислот:

Карбоновые кислоты. Среди кислот сильными восстановительными свойствами обладают муравьиная и щавелевая, которые окисляются до углекислого газа.
НСООН + HgCl 2 =CO 2 + Hg + 2HCl
HCOOH+ Cl 2 = CO 2 +2HCl
HOOC-COOH+ Cl 2 =2CO 2 +2HCl
Муравьиная кислота , кроме кислотных свойств, проявляет также некоторые свойства альдегидов, в частности, восстановительные. При этом она окисляется до углекислого газа. Например:
2KMnO4 + 5HCOOH + 3H2SO4 → K2SO4 + 2MnSO4 + 5CO2 + 8H2O
При нагревании с сильными водоотнимающими средствами (H2SO4 (конц.) или P4O10) разлагается:
HCOOH →(t) CO + H2O
Каталитическое окисление алканов:

Каталитическое окисление алкенов:

Окисление фенолов:


В заданиях категории С3 ЕГЭ особые трудности вызывают реакции окисления органических веществ перманганатом калия KMnO 4 в кислой среде, протекающие с разрывом углеродной цепочки. Например, реакция окисления пропена, протекающая согласно уравнению:
CH 3 – CH = CH 2 + KMnO 4 + H 2 SO 4 → CH 3 COOH + CO 2 + MnSO 4 + K 2 SO 4 + H 2 O.
Чтобы расставить коэффициенты в сложных уравнениях окислительно-восстановительных реакций, подобных этой, стандартная методика предлагает составить электронный баланс, но после очередной попытки становится очевидно, что этого недостаточно. Корень проблемы здесь кроется в том, что коэффициент перед окислителем, взятый из электронного баланса, необходимо заменить. Данная статья предлагает два способа, которые позволяют выбрать правильный коэффициент перед окислителем, чтобы, наконец, уравнять все элементы. Способ подстановки для замены коэффициента перед окислителем больше подходит тем, кто способен долго и кропотливо считать, поскольку расстановка коэффициентов этим способом может оказаться длительной (в данном примере понадобилось 4 попытки). Способ подстановки применяется совместно с методом "ТАБЛИЦА", который также подробно рассматривается в этой статье. Способ "алгебраический" позволяет не менее просто и надёжно, но гораздо быстрее заменить коэффициент перед окислителем KMnO 4 по сравнению со способом подстановки, однако имеет более узкую область применения. Способ "алгебраический" может быть использован только для замены коэффициента перед окислителем KMnO 4 в уравнениях реакций окисления органических веществ, протекающих с разрывом углеродной цепочки.
Скачать:
Предварительный просмотр:
Чтобы пользоваться предварительным просмотром создайте себе аккаунт (учетную запись) Google и войдите в него: https://accounts.google.com
По теме: методические разработки, презентации и конспекты
Расстановка коэффициентов в химических уравнениях
Преподаватель, являясь главным действующим лицом в организации познавательной деятельности учащихся, постоянно находится в поиске путей повышения эффективности обучения. Организация эффективного обуче...
4.5. Окисление алкенов
Реакции окисления алкенов целесообразно подразделить на две большие группы: реакции, в которых сохраняется углеродный скелет и реакции окислительной деструкции углеродного скелета молекулы по двойной связи. К первой группе реакций относятся эпоксидирование, а также гидроксилирование, приводящее к образованию вицинальных диолов (гликолей). В случае циклических алкенов при гидроксилировании образуются вицинальные транс - или цис -диолы. Другая группа включает озонолиз и реакции исчерпывающего окисления алкенов, приводящие к образованию различного рода карбонильных соединений и карбоновых кислот.
4.5.а. Реакции окисления алкенов с сохранением углеродного скелета
1. Эпоксидирование (реакция Н.А. Прилежаева, 1909 г)
Ациклические и циклические алкены при взаимодействии с перкислотами (надкислотами) RCOOOH в неполярной, индифферентной среде образуют эпоксиды (оксираны), поэтому сама реакция носит название реакции эпоксидирования.
Согласно современной номенклатуре ИЮПАК - трехчленный цикл с одним атомом кислорода носит название оксиран.
Эпоксидирование алкенов следует рассматривать как синхронный, согласованный процесс, в котором не участвуют ионные интермедиаты типа гидроксильного катиона ОН+ . Другими словами, эпоксидирование алкенов представляет собой процесс син -присоединения одного атома кислорода по двойной связи с полным сохранением конфигурации заместителей при двойной связи.

Для эпоксидирования был предложен механизм, характерный для согласованных процессов.

Т. к. атака двойной связи атомом кислорода надкислоты равновероятна с обеих сторон плоскости двойной связи, образующиеся оксираны представляют собой либо мезо -формы, либо смеси энантиомеров. В качестве эпоксидирующих агентов используются следующие перкислоты: пербензойная, м -хлорпербензойная, моноперфталевая, перуксусная, трифторперуксусная и пермуравьиная. Перкислоты ароматического ряда применяют в виде индивидуальных реагентов, тогда как перкислоты алифатического ряда - СН 3 СО 3 Н, CF 3 CO 3 H и НСО 3 Н не выделяют в индивидуальном виде, а используют после их образования при взаимодействии 30% или 90%-ного пероксида водорода и соответствующей карбоновой кислоты. Пербензойную и м -хлорпербензойную кислоты получают окислением соответственно бензойной и м -хлорбензойной кислот 70%-ной перекисью водорода в растворе метансульфокислоты или из хлорангидридов этих кислот и перекиси водорода.


Моноперфталевую кислоту получают подобным методом из фталевого ангидрида и 30%-ной перекиси водорода.

Первоначально для получения оксиранов (эпоксидов) использовались пербензойная или моноперфталевая кислоты:

В настоящее время для эпоксидирования чаще всего используют м -хлорпербензойную кислоту. В отличие от других перкислот она стабильна при хранении в течение длительного времени (до 1 года) и абсолютно безопасна при обращении. Выходы оксиранов, полученных при окислении ациклических и циклических алкенов м -хлорпербензойной кислотой в растворе хлористого метилена, хлороформа или диоксана, обычно довольно высоки.



Перкислоты часто генерируют прямо в реакционной смеси из 90% перекиси водорода и карбоновой кислоты в хлористом метилене.


Алкены с двойной связью, сопряженной с карбонильной группой или другим акцепторным заместителем, малоактивны и для их окисления лучше использовать более сильные окислители, такие как трифторперуксусная кислота, получаемая из ангидрида трифторуксусной кислоты и 90%-ной перекиси водорода в хлористом метилене. Простейший оксиран - окись этилена получают в промышленности окислением этилена кислородом в присутствии серебра, как катализатора.

2. анти -Гидроксилирование
Трехчленное кольцо оксиранов легко раскрывается под действием самых разнообразных нуклеофильных реагентов. Эти реакции подробно будут обсуждаться в разделе, посвященном ациклическим и циклическим простым эфирам. Здесь же будет рассматриваться только гидролиз оксиранов. Гидролиз оксиранов катализируется как кислотами, так и основаниями. В обоих случаях образуются вицинальные диолы, т. е. гликоли. При кислотном катализе в первой стадии происходит протонирование атома кислорода оксирана с образованием циклического оксониевого катиона, который раскрывается в результате нуклеофильной атаки молекулы воды:

Ключевой стадией в раскрытии кольца, определяющей скорость всего процесса, является нуклеофильная атака водой на протонированную форму оксирана. С точки зрения механизма этот процесс аналогичен раскрытию бромониевого иона при нуклеофильной атаке бромид-иона или другого нуклеофильного агента. С этих позиций стереохимическим результатом должно быть образование транс -гликолей при расщеплении циклических эпоксидов. Действительно, при кислотно-катализируемом гидролизе циклогексеноксида или циклопентеноксида образуются исключительно транс -1,2-диолы.

Таким образом, двухстадийный процесс эпоксидирования алкена с последующим кислотным гидролизом эпоксида суммарно соответствует реакции анти -гидроксилирования алкенов.
Обе стадии анти -гидроксилирования алкенов можно совместить, если алкен обрабатывать водной 30-70%-ной перекисью водорода в муравьиной или трифторуксусной кислоте. Обе эти кислоты являются достаточно сильными для того, чтобы вызвать раскрытие оксиранового цикла.

Раскрытие оксиранового кольца, катализируемое основанием, также приводит к образованию циклических транс -гликолей.

Следовательно, двухстадийный процесс эпоксидирования алкенов с последующим щелочным гидролизом эпоксидов также является реакцией анти -гидроксилирования алкенов.
3. син -Гидроксилирование
Некоторые соли и оксиды переходных металлов в высших степенях окисления являются эффективными реагентами син -гидроксилирования двойной связи алкена, когда обе гидроксильные группы присоединяются с одной и той же стороны двойной связи. Окисление алкенов перманганатом калия - один из старейших методов син -гидроксилирования двойной связи - продолжает широко использоваться, несмотря на свойственные ему ограничения. Цис -1,2-циклогександиол был впервые получен В.В. Марковниковым в 1878 году гидроксилированием циклогексена водным раствором перманганата калия при 0 0 С.

Этот метод в дальнейшем получил развитие в работах русского ученого Е.Е. Вагнера, поэтому син -гидроксилирование алкенов под действием водного раствора перманганата калия носит название реакции Вагнера. Перманганат калия является сильным окислителем, способным не только гидроксилировать двойную связь, но и расщеплять образующийся вицинальный диол. Для того, чтобы по возможности избежать дальнейшего расщепления гликолей, необходимо тщательно контролировать условия реакции. Выходы гликолей при этом обычно невелики (30-60%). Наилучшие результаты достигаются при гидроксилировании алкенов в слабощелочной среде (рН~8 9) при 0-5 0 С разбавленным 1%-ным водным раствором KMnO 4 .


Первоначально при окислении алкенов перманганатом калия образуется циклический эфир марганцевой кислоты, который немедленно гидролизуется до вицинального диола.

Циклический эфир марганцевой кислоты как интермедиат не был выделен, однако его образование следует из экспериментов с меченым 18 О перманганатом калия: оба атома кислорода в гликоле оказываются мечеными при окислении алкена KMn 18 O 4 . Это означает, что оба атома кислорода переходят от окислителя, а не из растворителя - воды, что находится в хорошем соответствии с предлагаемым механизмом.
Другой метод син -гидроксилирования алкенов под действием оксида осмия (VIII) OsO 4 был предложен Р. Криге в 1936 году. Тетраоксид осмия представляет собой бесцветное, летучее, кристаллическое вещество, хорошо растворимое в эфире, диоксане, пиридине и др. органических растворителях. При взаимодействии тетраоксида осмия с алкенами в эфире или диоксане образуется черный осадок циклического эфира осмиевой кислоты - осмат, который легко может быть изолирован в индивидуальном виде. Присоединение OsO 4 к двойной связи заметно ускоряется в растворе в пиридине. Разложение осматов до вицинальных гликолей достигается действием водного раствора гидросульфита натрия или сероводородом.

Выходы продуктов син -гидроксилирования алкенов в этом методе значительно выше, чем при использовании перманганата в качестве окислителя. Важным достоинством метода Криге является отсутствие продуктов окислительного расщепления алкенов, характерного для перманганатного окисления.


Тетраоксид осмия очень дорогой и труднодоступный реагент, к тому же он токсичен. Поэтому оксид осмия (VIII) используется при синтезе малых количеств трудно доступных веществ с целью получения наиболее высокого выхода диола. С целью упрощения син -гидроксилирования алкенов под действием OsO 4 была разработана методика, позволяющая использовать лишь каталитические количества этого реагента. Гидроксилирование алкенов осуществляется с помощью перекиси водорода в присутствии OsO 4 , например:

В заключение этого раздела приведем стереохимические отношения между алкеном цис - или транс -конфигурации и конфигурацией образующегося вицинального диола, который может быть цис - или транс -изомером, эритро - или трео -формой, мезо - или D,L -формой в зависимости от заместителей в алкене:

Аналогичные стереохимические отношения наблюдаются и в других реакциях син - или анти -присоединения по кратной связи водорода, галогенводородов, воды, галогенов, гидридов бора и др. реагентов.
Окислительно-восстановительные реакции с участием органических веществ
Склонность органических соединений к окислению связывают с наличием кратных связей, функциональных групп, атомов водорода при атоме углерода, содержащем функциональную группу.
Последовательное окисление органических веществ можно представить в виде следующей цепочки превращений:
Насыщенный углеводород→ Ненасыщенный углеводород → Спирт→ Альдегид (кетон) → Карбоновая кислота →CO 2 + H 2 O
Генетическая связь между классами органических соединений представляется здесь как ряд окислительно – восстановительных реакций, обеспечивающих переход от одного класса органических соединений к другому. Завершают его продукты полного окисления (горения) любого из представителей классов органических соединений.
Зависимость окислительно-восстановительной способности органического вещества от его строения:
Повышенная склонность органических соединений к окислению обусловлена наличием в молекуле веществ:
- кратных связей (именно поэтому так легко окисляются алкены, алкины, алкадиены);
- определенных функциональных групп , способных легко окисляться (–-SH, –OH (фенольной и спиртовой), – NH 2 ;
- активированных алкильных групп , расположенных по соседству с кратными связями. Например, пропен может быть окислен до непредельного альдегида акролеина кислородом воздуха в присутствии водяных паров на висмут- молибденовых катализаторах.
H 2 C═CH−CH 3 → H 2 C═CH−COH
А также окисление толуола до бензойной кислоты перманганатом калия в кислой среде.
5C 6 H 5 CH 3 +6KMnO 4 + 9H 2 SO 4 → 5C 6 H 5 COOH + 3K 2 SO 4 + 6MnSO 4 +14H 2 O
- наличие атомов водорода при атоме углерода, содержащем функциональную группу .
Примером является реакционная способность в реакциях окисления первичных, вторичных и третичных спиртов по реакционной способности к окислению.
Несмотря на то, что в ходе любых окислительно-восстановительных реакций происходит как окисление, так и восстановление, реакции классифицируют в зависимости от того, что происходит непосредственно с органическим соединением (если оно окисляется, говорят о процессе окисления, если восстанавливается – о процессе восстановления).
Так, в реакции этилена с перманганатом калия этилен будет окисляться, а перманганат калия – восстанавливается. Реакцию называют окислением этилена.
Применение понятия «степени окисления» (СО) в органической химии очень ограничено и реализуется, прежде всего, при составлении уравнений окислительно-восстановительных реакций. Однако, учитывая, что более или менее постоянной состав продуктов реакции возможен только при полном окислении (горении) органических веществ, целесообразность расстановки коэффициентов в реакциях неполного окисления отпадает. По этой причине обычно ограничиваются составлением схемы превращений органических соединений.
При изучении сравнительной характеристики неорганических и органических соединений мы знакомились с использованием степени окисления (с.о.) (в органической химии, прежде всего углерода) и способами ее определения:
1) вычисление средней с.о. углерода в молекуле органического вещества:
-8/3 +1
Такой подход оправдан, если в ходе реакции в органическом веществе разрушаются все химические связи (горение, полное разложение).
2) определение с.о. каждого атома углерода:
В этом случае степень окисления любого атома углерода в органическом соединении равна алгебраической сумме чисел всех связей с атомами более электроотрицательных элементов, учитываемых со знаком «+» у атома углерода, и числа связей с атомами водорода (или другого более электроположительного элемента), учитываемых со знаком «-» у атома углерода. При этом связи с соседними атомами углерода не учитывают.
В качестве простейшего примера определим степень окисления углерода в молекуле метанола.
![]()
Атом углерода связан с тремя атомами водорода (эти связи учитываются со знаком « – »), одной связью – с атомом кислорода (ее учитывают со знаком «+»). Получаем: -3 + 1 = -2.Таким образом, степень окисления углерода в метаноле равна -2.
Вычисленная степень окисления углерода хотя и условное значение, но оно указывает на характер смещения электронной плотности в молекуле, а ее изменение в результате реакции свидетельствует об имеющем место окислительно-восстановительном процессе.
Уточняем, в каких случаях лучше использовать тот или иной способ.
Процессы окисления, горения, галогенирования, нитрования, дегидрирования, разложения относятся к окислительно-восстановительным процессам.
При переходе от одного класса органических соединений к другому и увеличения степени разветвленности углеродного скелета молекул соединений внутри отдельного класса степень окисления атома углерода, ответственного за восстанавливающую способность соединения, изменяется.
Органические вещества, в молекулах которых содержатся атомы углерода с максимальными (- и +) значениями СО (-4, -3, +2, +3), вступают в реакцию полного окисления-горения, но устойчивых к воздействию мягких окислителей и окислителей средней силы .
Вещества, в молекулах которых содержится атомы углерода в СО -1; 0; +1, окисляются легко, восстановительные способности их близки, поэту их неполное окисление может быть достигнуто за счет одного из известных окислителей малой и средней силы . Эти вещества могут проявлять двойственную природу, выступая и в качестве окислителя , подобно тому, как это присуще неорганическим веществам.
При написании уравнений реакций горения и разложения органических веществ лучше использовать среднее значение с.о. углерода.
Например:
![]()
Составим полное уравнение химической реакции методом баланса.
Среднее значение степени окисления углерода в н-бутане:
Степень окисления углерода в оксиде углерода(IV) равна +4.
Составим схему электронного баланса:

Обратите внимание на первую половину электронного баланса: у атома углерода в дробном значении с.о. знаменатель равен 4, поэтому расчет передачи электронов ведем по этому коэффициенту.
Т.е. переход от -2,5 до +4 соответствует переходу 2,5 + 4 = 6,5 единиц. Т.к. участвует 4 атома углерода, то 6,5 · 4 = 26 электронов будет отдано суммарно атомами углерода бутана.
C учетом найденных коэффициентов уравнение химической реакции горения н-бутана будет выглядеть следующим образом:
Можно воспользоваться методом определения суммарного заряда атомов углерода в молекуле:
(4 C ) -10 …… → (1 C ) +4 , учитывая, что количество атомов до знака = и после должно быть одинаково, уравниваем (4 C ) -10 …… →[(1 C ) +4 ] · 4
Следовательно, переход от -10 до +16 связан с потерей 26 электронов.
В остальных случаях определяем значения с.о. каждого атома углерода в соединении, обращая при этом внимание на последовательность замещения атомов водорода у первичных, вторичных, третичных атомов углерода:


Вначале протекает процесс замещения у третичных, затем – у вторичных, и, в последнюю очередь – у первичных атомов углерода.
Алкены
Процессы окисления зависят от строения алкена и среды протекания реакции.
1.При окислении алкенов концентрированным раствором перманганата калия KMnO 4 в кислой среде (жесткое окисление) происходит разрыв σ- и π-связей с образованием карбоновых кислот, кетонов и оксида углерода(IV). Эта реакция используется для определения положения двойной связи.
а) Если двойная связь находится на конце молекулы (например, у бутена-1), то одним из продуктов окисления является муравьиная кислота, легко окисляющаяся до углекислого газа и воды:

б) Если в молекуле алкена атом углерода при двойной связи содержит два углеродных заместителя (например, в молекуле 2-метилбутена-2), то при его окислении происходит образование кетона , т. к. превращение такого атома в атом карбоксильной группы невозможно без разрыва C–C-связи, относительно устойчивой в этих условиях:

в) Если молекула алкена симметрична и двойная связь содержится в середине молекулы, то при окислении образуется только одна кислота:

Особенностью окисления алкенов, в которых атомы углерода при двойной связи содержат по два углеродных радикала, является образование двух кетонов:


2.В нейтральной или слабощелочной средах окисление сопровождается образованием диолов (двухатомных спиртов) , причем гидроксильные группы присоединяются к тем атомам углерода, между которыми существовала двойная связь:

В ходе этой реакции происходит обесцвечивание фиолетовой окраски водного раствора KMnO 4 . Поэтому она используется как качественная реакция на алкены (реакция Вагнера).
3. Окисление алкенов в присутствии солей палладия (Вакер-процесс) приводит к образованию альдегидов и кетонов:
2CH 2 =CH 2 + O 2 PdCl2/H2O → 2 CH 3 -CO-H
Гомологи окисляются по менее гидрированному атому углерода:
СH 3 -CH 2 -CH=CH 2 + 1/2O 2 PdCl2/H2O → CH 3 - CH 2 -CO-CH 3
Алкины
Окисление ацетилена и его гомологов протекает в зависимости от того, в какой среде протекает процесс.
а) В кислой среде процесс окисления сопровождается образованием карбоновых кислот:

Реакция используется для определения строения алкинов по продуктам окисления:

В нейтральной и слабощелочной средах окисление ацетилена сопровождается образованием соответствующих оксалатов (солей щавелевой кислоты), а окисление гомологов – разрывом тройной связи и образованием солей карбоновых кислот:

Для ацетилена:
1) В кислой среде:
H-C≡C-H KMnO 4, H 2 SO 4 → HOOC-COOH (щавелевая кислота)
3CH≡CH +8KMnO 4 H 2 O → 3KOOC-COOK оксалат калия +8MnO 2 ↓+ 2KOH+ 2H 2 O
Арены
(бензол и его гомологи)
При окисления аренов в кислой среде следует ожидать образования кислот, а в щелочной – солей.
Гомологи бензола с одной боковой цепью (независимо от ее длины) окисляются сильным окислителем до бензойной кислоты по α -углеродному атому. Гомологи бензола при нагревании окисляются перманганатом калия в нейтральной среде с образованием калиевых солей ароматических кислот.
5C 6 H 5 –CH 3 + 6KMnO 4 + 9H 2 SO 4 = 5C 6 H 5 COOH + 6MnSO 4 + 3K 2 SO 4 + 14H 2 O,
5C 6 H 5 –C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 = 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O,
C 6 H 5 –CH 3 + 2KMnO 4 = C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O.
Подчеркиваем, что если в молекуле арена несколько боковых цепей, то в кислой среде каждая из них окисляется по a-углеродному атому до карбоксильной группы, в результате чего образуются многоосновные ароматические кислоты:

1) В кислой среде:
С 6 H 5 -CH 2 -R KMnO 4, H 2 SO 4 → С 6 H 5 -COOH бензойная кислота + CO 2
2) В нейтральной или щелочной среде:
С 6 H 5 -CH 2 -R KMnO4, H2O/(OH) → С 6 H 5 -COOK + CO 2
3) Окисление гомологов бензола перманганатом калия или бихроматом калия при нагревании:
С 6 H 5 -CH 2 -R KMnO 4, H 2 SO 4, t ˚ C → С 6 H 5 -COOH бензойная кислота + R-COOH
4) Окисление кумола кислородом в присутствии катализатора (кумольный способ получения фенола):
C 6 H 5 CH(CH 3) 2 O2, H2SO4 → C 6 H 5 -OH фенол + CH 3 -CO-CH 3 ацетон
5C 6 H 5 CH(CH 3) 2 + 18KMnO 4 + 27H 2 SO 4 → 5C 6 H 5 COOH + 42H 2 O + 18MnSO 4 + 10CO 2 + K 2 SO 4
C 6 H 5 CH(CH 3) 2 + 6H 2 O – 18ē → C 6 H 5 COOH + 2CO 2 + 18H + | x 5
MnO 4 - + 8H + + 5ē → Mn +2 + 4H 2 O | x 18
Следует обратить внимание на то, что при мягком окислении стирола перманганатом калия КMnO 4 в нейтральной или слабощелочной среде происходит разрыв π -связи,образуется гликоль (двухатомный спирт). В результате реакции окрашенный раствор перманганата калия быстро обесцвечивается и выпадает коричневый осадок оксида марганца (IV).
Окисление же сильным окислителем – перманганатом калия в кислой среде – приводит к полному разрыву двойной связи и образованию углекислого газа и бензойной кислоты, раствор при этом обесцвечивается.
C 6 H 5 −CH═CH 2 + 2 KMnO 4 + 3 H 2 SO 4 → C 6 H 5 −COOH + CO 2 + K 2 SO 4 + 2 MnSO 4 +4 H 2 O
Спирты
Следует помнить, что:
1) первичные спирты окисляются до альдегидов:
3CH 3 –CH 2 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 = 3CH 3 –CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O;
2) вторичные спирты окисляются до кетонов:

3) для третичных спиртов реакция окисления не характерна.
Третичные спирты, в молекулах которых нет атома водорода при атоме углерода, содержащем группу ОН, в обычных условиях не окисляются. В жестких условиях (при действии сильных окислителей и при высоких температурах) они могут быть окислены до смеси низкомолекулярных карбоновых кислот, т.е. происходит деструкция углеродного скелета.
При окислении метанола подкисленным раствором перманганата калия или дихромата калия образуется CO 2 .
Первичные спирты при окислении в зависимости от условий протекания реакции могут образовать не только альдегиды, но и кислоты.
Например, окисление этанола дихроматом калия на холоду заканчивается oбразованием уксусной кислоты, а при нагревании – ацетальдегида:
3CH 3 –CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 = 3CH 3 –COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O,
Если три или более ОН-групп связаны с соседними атомами углерода, то при окислении иодной кислотой средний или средние атомы превращаются в муравьиную кислоту
Окисление гликолей перманганатом калия в кислой среде проходит аналогично окислительному расщеплению алкенов и также приводит к образованию кислот или кетонов в зависимости от строения исходного гликоля.
Альдегиды и кетоны
Альдегиды легче, чем спирты, окисляются в соответствующие карбоновые кислоты не только под действием сильных окислителей (кислород воздуха, подкисленные растворы KMnO 4 и K 2 Cr 2 O 7), но и под действием слабых (аммиачный раствор оксида серебра или гидроксида меди(II)):
5CH 3 –CHO + 2KMnO 4 + 3H 2 SO 4 = 5CH 3 –COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O,
3CH 3 –CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 = 3CH 3 –COOH + Cr 2 (SO 4) 3 + K 2 SO 4 + 4H 2 O,
CH 3 –CHO + 2OH CH 3 –COONH 4 + 2Ag + 3NH 3 + H 2 O
Особое внимание!!! Окисление метаналя аммиачным раствором оксида серебра приводит к образованию карбоната аммония, а не муравьиной кислоты:
HCH О + 4OH = (NH 4) 2 CO 3 + 4Ag + 6NH 3 + 2H 2 O.
Для составления уравнений окислительно- восстановительных реакций используют как метод электронного баланса, так и метод полуреакций (электронно-ионный метод).
Для органической химии важна не степень окисления атома, а смещение электронной плотности, в результате которого на атомах появляются частичные заряды, никак не согласующиеся со значениями степеней окисления.
Многие вузы включают в билеты для вступительных экзаменов задания по подбору коэффициентов в уравнениях ОВР ионно-электронным методом (методом полуреакций). Если в школе и уделяется хоть какое-то внимание этому методу, то, в основном при окислении неорганических веществ.
Попробуем применить метод полуреакций для окисления сахарозы перманганатом калия в кислой среде.
Преимущество этого метода заключается в том, что нет необходимости сразу угадывать и записывать продукты реакции. Они достаточно легко определяются в ходе уравнения. Окислитель в кислой среде наиболее полно проявляет свои окислительные свойства, например, анион MnO - превращается в катион Mn 2+ , легко окисляющиеся органические соединения окисляются до CO 2 .
Запишем в молекулярном виде превращения сахарозы:
![]()
В левой части не хватает 13 атомов кислорода, чтобы устранить это противоречие, прибавим 13 молекул H 2 O.
Левая часть теперь содержит 48 атомов водорода, они выделяются в виде катионов Н + :
Теперь уравняем суммарные заряды справа и слева:
Схема полуреакций готова. Составление схемы второй полуреакции обычно не вызывает затруднений:
Объединим обе схемы:
![]()
Задание для самостоятельной работы:
Закончите УХР и расставьте коэффициенты методом электронного баланса или методом полуреакций:
CH 3 -CH=CH-CH 3 + KMnO 4 + H 2 SO 4 →
CH 3 -CH=CH-CH 3 + KMnO 4 + H 2 О →
(CH 3) 2 C=C-CH 3 + KMnO 4 + H 2 SO 4 →
CH 3 -CH 2 -CH=CH 2 + KMnO 4 + H 2 SO 4 →
С H 3 -CH 2 -C≡C-CH 3 + KMnO 4 + H 2 SO 4 →
C 6 H 5 -CH 3 + KMnO 4 + H2O →
C 6 H 5 -C 2 H 5 + KMnO 4 + H 2 SO 4 →
C 6 H 5 - CH 3 + KMnO 4 + H 2 SO 4 →
Мои заметки:
Особое внимание учащихся следует обратить на поведение окислителя – перманганата калия КМnО 4 в различных средах. Это связано с тем, что окислительно-восстановительные ре акции в КИМах встречаются не только в заданиях С1 и С2. В заданиях СЗ, представляющих цепочку превращений органических веществ нередки уравнения окисления-восстановления. В школе часто окислитель записывают над стрелкой как [О]. Требованием к выполнению таких заданий на ЕГЭ является обязательное обозначение всех исходных веществ и продуктов реак ции с расстановкой необходимых коэффициентов.